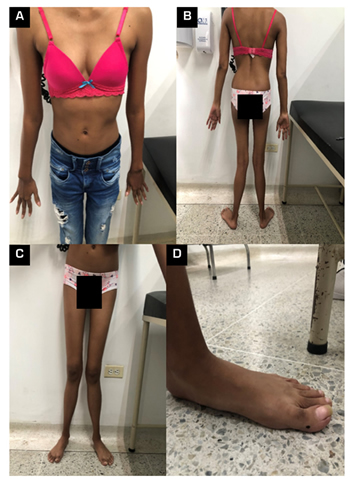
Fig. 1. Clinical photographs. A: upper limbs anterior view; B: upper and lower limbs posterior view; C: lower limbs anterior view; D: Left foot.
doi.org/10.20986/resed.2021.3930/2021
NOTA CLÍNICA
BONE PAIN MANAGEMENT WITH OPIOID MEDICATION IN A PATIENT WITH CAMURATI-ENGELMANN DISEASE: A CASE REPORT
MANEJO DEL DOLOR ÓSEO CON MEDICACIÓN OPIOIDE EN UNA PACIENTE CON ENFERMEDAD DE CAMURATI-ENGELMANN: REPORTE DE UN CASO
L. Arce Gálvez1
M. P. Grisales Gafaro1
K. Espinosa Soto1
C. Baena Álvarez1
1Department of Physical Medicine and Rehabilitation, Universidad del Valle, Cali, Colombia
1Department of Physical Medicine and Rehabilitation, Hospital Universitario del Valle, Evaristo García E.S.E. Cali, Colombia
1Department of Pain Medicine, Hospital Universitario del Valle, Evaristo García E.S.E. Cali, Colombia
ABSTRACT
Introduction: Camurati-Engelman Disease is a rare genetic sclerosing bone dysplasia with periosteal and endosteal thickening of the cortical of the long bones. It generates pain secondary to the reduction of the medullary canal that is usually controlled with corticosteroids and, in severe cases, with surgical decompression.
Case history: We present the case of a woman with a genetic diagnosis of Camurati-Engelman Disease with poor pain control with corticosteroid management and surgical procedures throughout her childhood and early adulthood. In whom optimal pain control was achieved with pain regimen with hydrocodone analgesic management. This is the first case described in the literature for adequate pain control using an opioid drug.
Discussion: CE disease is an extremely rare genetic entity with little more than 300 cases reported in the world. It is generated by an alteration in the gene for growth factor-beta 1 (TGF-B1); it has a varied clinical presentation that can begin with bone alterations accompanied by muscle weakness, joint angular alterations, headache, and nerve compressions. It has a differential diagnosis with some genetic entities that may present clinical similarity, but its morphological and radiological characteristics are distinctive. The usual management of bone pain generated by this entity is based on corticosteroids, in addition to losartan or surgical intervention aimed at reducing cortical changes. The intervention with opioid analgesics accompanied by a rehabilitation plan is not a frequent report, this being a case of success due to the refractoriness of the symptoms in a patient with chronic pain, with a positive impact on her functionality and quality of life.
Conclusion: It is considered that analgesic management with opioids may be a treatment option in patients with Camurati-Engelman disease refractory to corticosteroid management and surgical interventions.
Key words: Opioid, pain treatment, Camurati-Engelmann, progressive diaphyseal dysplasia, TGFB-1
RESUMEN
Introducción: La enfermedad de Camurati-Engelman (CE) es una displasia ósea esclerosante rara, de causa genética. Se presenta con engrosamiento perióstico y endóstico de la cortical de los huesos largos. Genera dolor secundario a la reducción del canal medular, que habitualmente se controla con corticoides y en casos severos, con descompresión quirúrgica.
Historia del caso: Presentamos el caso de una mujer con diagnóstico genético de enfermedad de Camurati-Engelman, con mal control del dolor, con manejo de corticosteroides y procedimientos quirúrgicos a lo largo de su niñez y adultez temprana. Se logró un control óptimo del dolor con un régimen con manejo analgésico con hidrocodona. Este es el primer caso descrito en la literatura de un adecuado control del dolor con un medicamento opioide.
Discusión: La enfermedad de CE es una entidad genética extremadamente rara, con poco más de 300 casos reportados en el mundo. Se genera por una alteración en el gen del factor de crecimiento beta 1 (TGF-B1). Tiene una presentación clínica variada que puede iniciar con las alteraciones óseas acompañado de debilidad muscular, alteraciones angulares articulares, cefalea y compresiones nerviosas. Tiene diagnóstico diferencial con algunas entidades genéticas que pueden presentar similitud clínica, pero su característica morfológica y radiológica es distintiva. El manejo usual del dolor óseo generado por esta entidad se basa en corticoesteroides, además de losartán o intervenciones quirúrgicas orientadas a disminuir los cambios corticales. La intervención con analgésicos opioides, acompañada de un plan de rehabilitación, no es un reporte frecuente, siendo este un caso de éxito ante la refractariedad de los síntomas en una paciente con dolor crónico, impactando de manera positiva en su funcionalidad y calidad de vida.
Conclusión: Se considera que el manejo analgésico con opioides puede ser una opción de tratamiento en pacientes con enfermedad de Camurati-Engelman refractaria al manejo con corticoides e intervenciones quirúrgicas.
Palabras clave: Opioide, tratamiento del dolor, Camurati-Engelmann, displasia diafisaria progresiva, TGFB-1
Recibido: 01-06-2021
Aceptado: 20-11-2021
Correspondencia: Leonado Arce Gálvez
leonardo.arce@correounivalle.edu.co
INTRODUCTION
Camurati-Engelmann Disease (CED) or progressive diaphyseal dysplasia is a rare autosomal dominant sclerosing bone dysplasia with around 300 cases described worldwide (1). Its clinical presentation is around 13 years with gait disturbances caused by an increase in the polygon of support and a waddling gait, headache, pain in the extremities, fatigue, and proximal muscle weakness (1,2). From the radiological point of view, it is characterized by cortical thickening of the at the diaphysis in long bones, secondary to blast activity in the periosteal and endosteal surfaces, producing a narrowing of the medullary canal; additionally, it can be observed compromise at the level of the skull and the pelvis (1,3,4). The treatment of this disease is based on corticosteroids at high doses due to its theoretical induction of osteoclastic activity and, in severe cases, surgical decompression of the medullary canals (3,4). In this case, we present a patient with inadequate response to traditional management, who improved with opioid analgesia, which has not been previously described in the literature available for this disease.
CASE HISTORY
We present a 26-year-old female patient (Figure 1), who began at four years of age with somatic nociceptive pain in the lower limbs, associated with weight loss and waddling. Due to these symptoms, she consulted primary care, where they took radiographs of their lower limbs that reported blast density at the intramedullary level in both tibiae. Initially, radiographic follow-up and management with paracetamol 15 mg/kg were indicated for pain control, which achieved partial control of pain, allowing her to perform age-appropriate activities. After two years of follow-up, she presented worsening pain. Her treating physician requested new x-rays of the tibiae that report a marked increase in spinal sclerosis in the tibiae. With this result, it was decided to take a bone biopsy that reported sclerosis compatible with progressive diaphyseal dysplasia. Given these findings, they conducted a genetic panel that reported a mutation of the TGFB1 gene, compatible with CED. Management was started with steroids and paracetamol for pain control.
Fig. 1. Clinical photographs. A: upper limbs anterior view; B: upper and lower limbs posterior view; C: lower limbs anterior view; D: Left foot.
At twelve years of age, the symptoms worsened, presenting headache and nociceptive pain in all four extremities, especially in the lower limbs. For this reason, a bone scan was performed, which showed intense diffuse osteoblastic activity that compromised the skull, humerus, and femurs symmetrically. It was decided to increase the dose of steroids, but the objectives in pain control were not achieved. Therefore, the orthopedic department evaluated the patient, where they considered performing a surgical decompression with recanalization of the tibiae, thus generating clinical improvement for more than six years.
At the end of her 18 years, her symptoms worsen again, with difficult pain control despite high doses of steroids. New radiographs were requested that reported severe spinal obstruction and bone thickening of the diaphysis of both femurs and tibiae, as well as a bone scan with the progression of osteoblastic activity predominantly of proximal and distal thirds of long bones, including femurs, tibiae, humerus, radii, ulnas, and axial skeleton involvement. With these imaging findings, it was decided to perform a new bone recanalization and continue the steroids. Despite these therapeutic efforts, six months after the last intervention, the pain in all four extremities worsened, adverse effects to the prolonged use of steroids appeared, leading to the suspension of this medication, and there were no surgical treatment options available, so the patient referred for evaluation in the pain clinic.
In the clinical assessment of pain, her inadequate response to conventional management for previously tried CED was considered (Table I) and the persistence of somatic nociceptive pain. Under this case scenario, she was considered a candidate for multimodal management with paracetamol, opioid medication, and gabapentinoid-type adjuvant, starting paracetamol/hydrocodone 500 mg / 5 mg 1 tablet every 8 hours and pregabalin 75 mg per day. Additionally, non-pharmacological measures were prescribed for pain control with physical, occupational, and psychological therapy. The patient manifested an increase in daytime symptoms such as dizziness and drowsiness with the use of the gabapentinoid, so it was suspended, and the paracetamol/hydrocodone combination was continued. In the first follow-up of 15 days and the subsequent ones for more than eight years, adequate pain control was achieved without adding new drugs, increasing the dose, or performing a new surgical intervention.
Table I. Used medications, prior to pain clinic
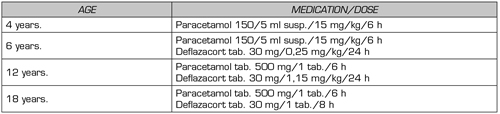
ml: milliliter. kg: kilogram. h: hour. susp.: suspension. tab.: tablets.
DISCUSSION
CED is an infrequent heterozygous mutation genetic entity, with just over 300 cases have been reported worldwide and unknown prevalence (5,6). One of its most frequent clinical characteristics is bone pain secondary to diaphyseal hyperostosis of the skull and long bones, a pathognomonic feature (7). It can also be accompanied by muscle weakness, fatigue, exophthalmos, headache, joint angular changes, alteration in bone mineral density, auditory symptoms, and various neurological manifestations due to bone compression of the nerves (3).
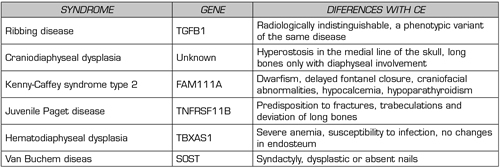
The diagnosis is based on the clinical manifestations and radiological findings of the extremities and the bone scan, which can be helpful. Typically, there is a fusiform cortical thickening of the diaphysis, which may extend to the metaphysis, but never to the epiphysis, and a narrow medullary canal (5,8). The study can be complemented with a specific genetic test for the gene that encodes growth factor beta-1 (TGF-B1) located in the 19q13.1-13.38 region (5). Some entities should be considered as diagnoses such as Ribbing’s disease, craniodiaphyseal dysplasia, Kenny-Caffey syndrome type 2, juvenile Paget’s disease, diaphyseal dysplasia with anemia, and van Buchem’s disease, with clinical similarities but differences in radiological findings and genetic tests oriented to different locations (1) (Table II).
Table II. CE differential diagnosis
Patients with CED generally present their first symptoms in childhood around four years of age, beginning with waddling gait and fat accumulation in the extremities frequently confused with muscular dystrophies (2). Over the years, the symptoms progress, compromising the patient’s functionality and leading to multiple complications. A series of annual follow-up evaluations should be carried out, including a comprehensive neurological clinical examination, blood count, blood pressure measurement, audiometry, bone densitometry, and monitoring of signs and symptoms of endocranial hypertension in patients with cranial hyperostosis (1).
Pain management is one of the essential aspects of the comprehensive approach to the patient with CED. The literature classically describes steroid interventions using prednisolone or deflazacort (5). Its effectiveness is attributed to the anti-inflammatory action and the reduction of bone mineral. Nevertheless, the secondary effects of metabolic predominance are the main obstacle to its use (8). A pharmacological option in patients who cannot use steroids is losartan (9). It is believed its effect is related to its anti-TGF-B1 action, but its effectiveness has only been reported in anecdotal cases (9). Bisphosphonates and calcitonin have a controversial role. In small case series, increased pain and bone scan activity has been reported, so these medications do not indicate this disease (8). Other pharmacological interventions with anti-inflammatories and analgesics reported in the literature have not impacted pain or the natural history of the disease (8).
Surgical treatment is used in long bone hyperostosis. Cases managed with cleaning the medullary canal or osteotomies have been described, although there is a high incidence of recurrence during the disease (10). In the case of hyperostosis of the skull that compresses the outlet of the cranial nerves, it can be managed with the surgical release but with a high chance of recurrence, which has raised different interventions, especially in the auditory nerve with cochlear implants, being still controversial intervention in CED (11).
Pharmacological intervention with opioid drugs is highly effective in controlling severe pain in multiple entities, including oncological and degenerative entities (12). Its use worldwide has increased, generating concern about its recreational use and the risk of addiction; however, in patients with adequately cataloged pain and a clear clinical strategy, they are a valuable and safe tool. In CED, they have been used occasionally without reaching the desired objectives, which has favored multiple pharmacological and surgical interventions that in the long term do not resolve the symptoms or have a high metabolic cost. In this patient’s case, an intervention with a hydrocodone-type opioid and a gabapentinoid analgesic adjuvant was proposed, extrapolating the recommendations for other pathologies for uncontrolled somatic nociceptive pain.
In the follow-up, the gabapentinoid was withdrawn due to low tolerance and secondary events such as drowsiness and dizziness, evidenced in patients with CED and other types of pathologies (13-15). By continuing the opioid, an adequate symptom control, and no dose increase or adjustment was required to achieve a sustained effect over time. Used appropriately, opioid medication use should be considered an effective and safe alternative for patients with CED, which might be part of the therapeutic options for physicians who treat this type of disease.
CONCLUSIONS
CE disease is an infrequent entity characterized by bone pain; steroids have been the treatment of choice, supplemented with surgical interventions. In our case, it is highlighted that the patient achieves adequate control with opioid medication; in the literature, there are no previous reports of cases with pain control with this type of analgesic, which offers the possibility of having an additional option of management and future studies to generate recommendations for use.
ACKNOWLEDGMENTS
To the Hospital Universitario del Valle Evaristo García E.S.E.
ETHICAL CONSIDERATION
The patient gave his consent for the use of clinical and paraclinical information, it was authorized by the ethics committee of the Hospital Universitario del Valle, Cali, Colombia.
CONFLICT OF INTEREST
We declare that we have no conflict of interest.
FUNDING
The authors did not receive funding to carry out this report.
REFERENCES