
DOI: 10.20986/resed.2020.3814/2020
SPECIAL ARTICLE
Clinical pathophysiology in patients with sickle cell disease: the transition from acute to chronic pain
Fisiopatología clínica en pacientes con enfermedad de células falciformes: la transición del dolor agudo al crónico
B. Mugabure Bujedo1,2
S. González Santos1
A. Uría Azpiazu1
A. Osorio López1,3
1Departamento de Anestesiología, Cuidados Críticos y Medicina del Dolor Perioperatoria. Hospital Universitario de Donostia. San Sebastián, Spain
2Unidad del Dolor, Manejo del Dolor Crónico. Hospital Universitario de Donostia. San Sebastián, Spain
3MIR Anestesiología. Hospital Universitario de Donostia. San Sebastián, Spain
ABSTRACT
Patients with sickle cell disease (SCD) suffer from severe pain that often begins in childhood and increases in severity over the course of a lifetime, leading to hospitalization and poor quality of life over the years. A unique feature of SCD is vase-occlusive crises (VOC) characterized by recurrent and unpredictable episodes of acute pain. Microvascular occlusion during a VOC results in decreased oxygen supply to the periphery and injury from ischemia and subsequent reperfusion, inflammation, oxidative stress and endothelial dysfunction, all of which can perpetuate a harmful pain-causing microenvironment. On the other hand, in addition to episodic acute pain, SCD patients also report chronic pain, defined as almost daily pain over a 6-month period associated to either sicologic or social morbidities. They may be due to chronic lesions such as skin ulcers, avascular bone necrosis or infarctions in various organs. In addition, central sensitization appears to be directly involved in the chronicity of pain and there is a clearly under-diagnosed and under-treated component of neuropathic pain. Current treatment of moderate to severe pain in SCD is based primarily on opioids; either as an oral quick release outpatient or in the form of patient-controlled intravenous analgesia in the hospital. However, long-term opioid use is associated with multiple side effects. This review presents the latest advances in the understanding of the pathology of pain in SCD and describes objectives based on mechanisms that may help to develop new therapeutic and/or preventive strategies to improve pain in SCD.
Key words: Analgesia, sickle cell disease, acute pain, chronic pain syndrome, opioids
RESUMEN
Los pacientes con enfermedad de células falciformes (ECF), también denominada drepanocítica, sufren un dolor intenso que suele comenzar durante la infancia y aumenta su gravedad a lo largo de la vida, lo que lleva a la hospitalización y a una mala calidad de vida a lo largo de los años. Una característica única de la ECF son las crisis vaso-oclusivas (CVO), caracterizadas por episodios recurrentes e impredecibles de dolor agudo. La obstrucción microvascular durante una CVO provoca una disminución del suministro de oxígeno a la periferia y una lesión por isquemia y posterior reperfusión, inflamación, estrés oxidativo y disfunción endotelial, todo lo cual puede perpetuar un microambiente nocivo que provoca dolor. Por otro lado, además de los dolores agudos episódicos, los pacientes con ECF también padecen dolor crónico, definido como dolor casi diario durante un periodo de 6 meses, asociado a trastornos psicosociales. Pueden deberse a lesiones crónicas como úlceras cutáneas, necrosis avascular ósea o infartos en diversos órganos. Asimismo, la sensibilización central parece estar directamente involucrada en la cronicidad del dolor y existe un componente de dolor neuropático claramente infradiagnosticado e infratratado. El tratamiento actual del dolor moderado a intenso en la ECF se basa principalmente en la administración de los opioides, vía oral, de liberación rápida ambulatoria o en forma de analgesia controlada por el paciente vía intravenosa intrahospitalaria. Sin embargo, el uso de opioides a largo plazo está asociado con múltiples efectos secundarios. Esta revisión presenta los últimos avances en la comprensión de la fisiopatología del dolor en la ECF y se describen los mecanismos subyacentes que pueden ayudar a desarrollar nuevas estrategias terapéuticas y/o preventivas para mejorar el dolor en la ECF.
Palabras clave: Analgesia, enfermedad de células falciformes, dolor agudo, síndrome de dolor crónico, opioides
Correspondence: Borja Mugabure Bujedo
mugabure@yahoo.es
Received: 17-04-2020
Accepted: 07-07-2020
INTRODUCTION
Sickle cell disease (SCD) is an autosomal recessive hemoglobin disorder characterized by homozygous or heterozygous mutations in beta-globin genes. Replacing glutamic acid with valine in position 6 generates an abnormal hemoglobin called Hb S (1). It affects approximately 100,000 people in the United States and 25 million people worldwide, with preference for the African-American population (2,3). Vaso-occlusive crises are one of the characteristic manifestations of the disease and involve frequent visits to emergency services and hospital admissions, which are estimated to cost $2400 million annually in the United States (4). Evidence of opioid use patterns in this patient population is quite limited, and there is still controversy over the opioid dose that patients with SCD use in daily lives. Many studies show that patients tend to use more opioids than many other chronic pain conditions, while some studies showed a relatively low dose of opioids used in this patient population (5,6).
The frequency and severity of pain of SCD increases with age, and a subset of children develop chronic pain syndrome in adolescence and adulthood, as well as episodes of recurrent acute pain. It has been reported that 30-40% of adolescents and adults with SCD have chronic pain daily (7,8) and continuous episodes of acute pain overlap with chronic pain, all of which seriously affect health-related quality of life (9,10). These subjects have a life expectancy of about 50 years due to serious complications, such as acute chest syndrome, infarctions in long bones and in various organs such as the kidney, as well as infections secondary to encapsulated bacteria, such as Streptococcus pneumoniae, therefore, the corresponding vaccines and prophylactic antibiotic treatments are crucial. Hydroxyurea (HU) and L-glutamine are approved by the Food and Drugs Administration (FDA) and are disease-modifying medications for preventing episodes of acute pain. HU has demonstrated a protective ability in SCD, due to the increase in Hb F (fetal hemoglobin) by reducing the enzyme ribonucleotide reductase, improving up to 50% of VOC (11,12). Despite the significant morbidity that pain causes in people with SCD, the underlying biology of pain, which produces both acute and chronic pain, is not fully defined.
Classically, the pain of SCD has been attributed to nociceptive or inflammatory pain resulting from repeated vascular occlusion induced by sickle red blood cells, chronic ischemia-reperfusion injury, and subsequent inflammation (13,14). However, it is now understood that sickle cell pain is very complex, multifactorial and variable, resulting in nociceptive, inflammatory and neuropathic pain. People with SCD often report significant chronic pain (7,8) in multiple sites along the body (15) that cannot be attributed to an identifiable cause or tissue injury, such as bone avascular necrosis, leg ulcers, or microembolism (16). In addition, although opioids and non-steroidal anti-inflammatory drugs are the backbone of the disease’s pain treatment, they are often ineffective and only achieve partial pain relief. This pain pattern presents clinical challenges of treatment approach that have led to research into alternative pathways in the etiology of pain in sickle cells that are outside the abnormality of red blood cells. It is important that many of these studies have focused on the contribution of central and peripheral nervous system abnormalities to pain along with altered mechanisms in the pain pathways. Because of these studies, central sensitization has emerged as one of the underlying components of the pain experience in SCD. Chronic pain in this case may be associated with activation of glia cells and release of inflammatory mediators in the central nervous system (CNS).
To try to clarify these doubts, a search in Ovid, Medline, Embase and Cochrane Databases was performed for this narrative review to identify all articles published up to May 2020. The keywords analgesia, sickle cell disease, acute pain, chronic pain syndrome and opioids were searched. Systematic or narrative reviews, meta-analyzes, or Cochrane reviews about the SCD have been compiled. The analgesic agents that have not undergone such a systematic combination of data were individually evaluated for narrative review. Relevant studies addressing the pathophysiology of pain were obtained through a search in Ovid and Medline and reviewed individually to identify any studies that might have been lost in the main search. Additional material was retrieved by manually reviewing relevant article references identified with the highest level of impact and prior citations. All the finally included references were chosen by the authors to focus of the revision on the terms of interest.
PATHOPHYSIOLOGY
Pain transmission circuits
Neurobiology of pain is known to involve transduction, transmission, modulation, and perception of pain through the somatosensory and limbic systems (17,18). The first step is the conversion of a harmful stimulus (including mechanical, chemical, thermal and other algogenic agents) into an electric nerve impulse in the form of an action potential. The latter is generated by the influx of positively charged ions into an axon through voltage-dependent sodium channels through the neuronal membrane. The action potentials are spread through the primary afferent nerve fibers whose cell bodies located in the dorsal root ganglion (DRG) of the spinal cord are connected to second-order neurons in the dorsal horn of the spinal cord. Second-order neurons respond to nociceptive inputs of the A-d and C fibers and non-nociceptive inputs of the A-b fibers. A-d fibers respond to mechanical and thermal stimuli and are responsible for acute pain, such as the one that perhaps evokes a VOC. These fibers instantly activate second-order neurons. C fibers respond to a wide range of stimuli, including thermal, mechanical, and chemical stimuli that produce long-lasting, slow deaf pain, and trigger second-order neurons by maintaining depolarization status over a long period of time. A-b fibers transmit the sensation of touch and also proprioception, are low-threshold polymodal receptors. Therefore, activation of these fibers can suppress pain (such as massage of a painful area to relieve pain), and they are essential to regulate the entrance door at the DRG level, according to Melzack and Wall Gate theory described in 1965. The arrival of information through primary nociceptive afferent fibers is therefore as important as through non-nociceptive afferent fibers (19).
Second-order neurons in the spinal cord allow the transmission of nociceptive sensation or touch to the brain. These neurons are activated by glutamate and substance P (SP). The spinal dorsal horn acts as a “gate” for the convergence of peripheral fibers and descending fibers of the higher brain centers. Impulses transmitted from the periphery to this locus may be modulated by inputs from the descending neuronal fibers, as well as by the large A-b fibers, after a complex interaction with the excitatory and inhibitory neurotransmitters and the GABAergic interneurons. Finally, the descending system of pain modulation that originates in the higher brain centers plays a fundamental role in the perception of the afferent signal by inhibiting or amplifying nociceptive inputs through the neurotransmitters serotonin, dopamine, endogenous opioids, g-amino butyric acid, glycine and others. Modulated signals are processed in areas of the brain responsible for sensory perception and stimulation of affect, emotion and memory. The nature and severity of the conscious perception of pain depend on complex processing in different areas of the brain, including the somatosensory cortex, the prefrontal cortex, the amygdala and the anterior cingulate cortex. Recently, attention has focused on perception-based components of nociception because of their ability to reduce pain through nonpharmacological behavioral techniques, such as guided imagery, mindfulness and relaxation training, hypnosis, and cognitive behavioral therapy (20).
Alterations described in the SCD
Sickle cell disease ranges from a local mutation to systemic dysfunction and pain. The circulation of sickle-cell red blood cells leads to multiple pathophysiological problems, including, but not limited to, hemolysis, hypoxia/reperfusion, ischemia, excessive inflammation and free hemoglobin, by rupture of red blood cells, vascular dysfunction, organ damage and vascular occlusion. Each can contribute to a harmful microenvironment evoking the nociceptive mechanisms of pain. Emerging data have identified several cellular and molecular targets contributing to nociceptive activity in sickle cell anemia. These targets may be located in the periphery and/or CNS, suggesting that the microenvironment due to cell and/or tissue damage generated by SCD can trigger pain transmission from the periphery and directly influence the CNS. The mechanisms of nociception are complex, since they involve peripheral and central neural activity involving various processes (13):
Finally, peripheral and/or central sensitization may occur in response to ongoing nociceptive stimuli, resulting in a reduction in the threshold triggering potential leading to pain generation with harmless stimuli (allodynia). Nerve impulses are transmitted orthodromically from the periphery to the spinal cord, but under sustained activation for both the duration and intensity of the stimulus, they can travel in an antidromic direction, releasing neurotransmitters such as substance P (SP) in the periphery, conditioning the appearance of secondary hyperalgesia. In addition, the release of excitatory neuropeptides can also occur in the periphery from axonal nerve endings activated by axonal reflex, which favors nociceptive sensitization (22). Given the genetic nature of SCD, an ongoing harmful microenvironment full of algogenic factors can induce nociceptive mechanisms during childhood and maintain activation until adulthood if the disease remains uncontrolled. This results in peripheral and central sensitization resulting in chronic pain refractory to classical therapy. Therefore, recently, attention has focused on perception-based components of pain because of their ability to reduce pain through nonpharmacological behavioral techniques, such as imagery, mindfulness and relaxation training, hypnosis and cognitive behavioral therapy (23).
Neurogenic inflammation
Activated C fibers release vasoactive and proinflammatory neuropeptides, including SP and calcitonin gene-related peptide (CGRP), which stimulate arteriolar vasodilation and vascular leakage. In addition, inflammatory mediators are released, from the immune cells present in the altered area, from the endothelial cells and Schwann cells themselves, and finally from the tissue cells, which contribute to neurogenic inflammation. The orthodromic transmission of SP and CGRP to second-order neurons also contribute to central sensitization and lead to peripheral sensitization by being antidromically released to peripheral nerve fibers. These vasoactive and proinflammatory neuropeptides can promote vascular dysfunction by increasing vascular permeability, as well as activation of peripheral nociceptors, thus contributing to multiple characteristics of SCD, including dactylitis and pain. Given the low ability of standard anti-inflammatory agents to improve neurogenic inflammation, it is likely that new pharmacological agents targeting this pathway will need to be developed and tested in SCD. This is a general trend in a large number of diseases refractory to the use of traditional and systemic drugs. Therefore, therapeutic lines are opened that basically study the regulatory treatments of local inflammatory processes (24,25).
Central sensitization
Chronic pain in SCD is a pathophysiological entity different from pain in acute episodes because the initial origin of the lesion is usually not relevant once the sensory pathways become hyperexcitable. This process of pain chronification begins with peripheral sensitization of nociceptors by activation of the ionic channels by the proinflammatory substances, activating the vanilloid 1 (VRPT1) and sodium (Na+) receptor channels, and the subsequent impairment of the descending inhibitory control mediated by inhibitory interneurons (interneuronal death is associated with the process of excitotoxicity mediated by so-called dark neurons), which modifies the balance toward a state of neuronal hyperexcitability. This hyperexcitability involves a greater sensitivity to low-intensity harmful stimuli (hyperalgesia) or even non-painful stimuli (allodynia). In both cases, as seen in the laboratory with mice with sickle-shaped features, responses are perceived as high-intensity painful stimuli (26,27,28).
In relation to the above, electrophysiological records in a transgenic model of sickle-cell mice (BERK) revealed an increase in excitability of nociceptive neurons in the dorsal horn of the spinal cord, demonstrated by the increase in receptive fields, the exaggerated rate of spontaneous activity, increased responsiveness and prolongation of post-mechanical stimulus shocks (temporal summation phenomenon), and lower mechanical threshold compared to BERK controls (29). Activation of signaling pathways involved in neuronal hyperexcitability was observed, including mitogen-activated protein kinases, c-jun kinase, p42/p44 extracellular receptor kinase, and p38, as well as signal transducer and activator of transcription 3, on the spinal cords of mice with sickle-shaped cells. They also found high reactive oxygen species (ROS), SP, and microglial and astrocytic cells activated with increased glial fibrillary acid protein (GFAP) in the dorsal horn of the spinal cord of sickle mice (30). In human studies, increased GFAP circulation was reported in children with SCD compared to controls. Therefore, increased oxidative stress and neuroimmune interactions can contribute to neuronal hyperexcitability and chronic pain, pathways that can provide targets for novel therapeutic strategies (31).
In patients with SCD, quantitative sensory testing (QST) showed larger sensitivity to mechanical and thermal stimuli (heat and cold), suggesting central sensitization (32,33,34). Neuroimaging studies in patients with SCD showed larger pro-nociceptive connectivity and reduced anti-nociceptive connectivity in the default mode network (DMN) of patients with longer hospital stays related to pain compared to those with shorter stays (35). Electroencephalography recordings made simultaneously with functional magnetic resonance imaging in patients with SCD clarified neurophysiological manifestations of resting-state networks, including DMN. Increased electroencephalography activity was observed in patients during rest in pain-processing regions. This is consistent with functional magnetic resonance imaging findings of reduced DMN activity and increased activity in pain-processing regions compared to control subjects. Based on previous findings, a significant reduction in voxels was demonstrated in patients with SCD in the executive control network involved in cognitive functions, including perception and decision-making. Poor cognitive function and impairments in the executive control network have been found in patients with SCD (36,37).
Therefore, findings in sickle-cell mice and patients with SCD suggest peripheral sensitization of nociceptors, hyperexcitability of central neurons and activation of signaling mechanisms, as well as alterations in brain function and activity suggesting central sensitization. It is speculated that central sensitization can contribute to variability in pain intensity and that it is medically refractory to classic treatments for the acute episode. The possible therapeutic implication of these observations is that centrally-mediated pain states respond better to serotonin-norepinephrine reuptake inhibitors and central mechanism modulators, whereas peripheral and nociceptive pain states may respond better to opioids and non-steroidal analgesics. This different response is probably related to activation of glia cells in the chronification process (38).
TRANSITION FROM ACUTE PAIN TO CHRONIC PAIN
The etiology of acute pain episodes in SCD is complex and is assumed to be due to multiple disease-specific factors, such as oxidative stress, ischemia-reperfusion injury, vascular occlusion, and neuroinflammation. However, in clinical practice, episodes of acute pain are labeled as “episodes of acute vaso-occlusive pain” regardless of diagnostic studies to identify the presence of neuropathic pain and other pain phenotypes. Consequently, patients are treated non-specifically with acetaminophen, non-steroidal anti-inflammatory drugs, and opioids, which do not necessarily target the underlying mechanisms causing acute pain. This justifies further studies to elucidate the complex biology of acute pain in SCD, with the ultimate aim of developing a “lexicon of pain in sickle cell disease” that effectively links pain sub-phenotypes and their underlying pathophysiology with specific therapeutic interventions. Abrupt onset of episodes of acute pain usually occurs in the back, extremities, chest, and abdomen. Temporarily associated pain triggers include acute infections, dehydration, asthma, cold temperatures, and onset of menstruation, but often no trigger is identified (39,40,41,42). Episodes of acute pain may begin in the first few months of life, increase their frequency with age, and may contribute to the development of a chronic pain syndrome. As discussed above, acute pain is caused by recurrent vaso-occlusion of sickle cell cells with consequent ischemia-reperfusion injury, while chronic pain is likely to be driven by other reasons, such as central nervous system sensitization and glia cell hypertrophy (43).
Diagnosis of chronic pain syndrome in SCD is difficult and includes many biological, psychological, and sociological risk factors. Traditionally, chronic pain is defined as pain that persists for at least 3 to 6 months after the normal healing time of a tissue. However, evidence-based consensus diagnostic criteria have been established for chronic pain syndrome in SCD, as these patients have recurrent pain episodes throughout their life since childhood, so the following definition has been agreed: “episodes of ongoing pain on most days over the past 6 months, either in a single location or in multiple locations on the body.” Biopsychosocial mechanisms and chronic pain conditions in SCD are described in a recent comprehensive report of the collaborative working group representing various organizations related to pain and addiction, the U.S. Food and Drug Administration and the American Pain Society Pain Taxonomy AAPT (16). The classification proposal is for a single pain condition, which we label as chronic SCD pain, with 3 subtypes: 1) chronic SCD pain without contributory disease complications; 2) chronic SCD pain with contributory disease complications; and 3) chronic SCD pain with mixed pain types (Table 1).
Therefore, the evaluation of the pain of SCD should determine whether the pain is acute, chronic, unrelated to SCD or all three. A detailed medical record with the affected individual is required to distinguish between different types of pain. Pain associated with “excessive movement syndrome,” which is defined as pain caused by repetitive movements in daily activities, may be misunderstood and treated as acute disease pain, chronic pain syndrome, or an episode of long-term acute pain. The temporal association of the new onset of pain, together with the location and type of pain, can help the affected subject and the physician distinguish the etiology of pain. The data show that individuals with SCD use descriptions suggesting both the origins of nociceptive pain (crushing, drilling, tearing) and neuropathic pain (cold, hot, stabbing, cramping). It is obvious, therefore, that obtaining the characteristics of pain may improve the treatment of SCD-associated pain (44).
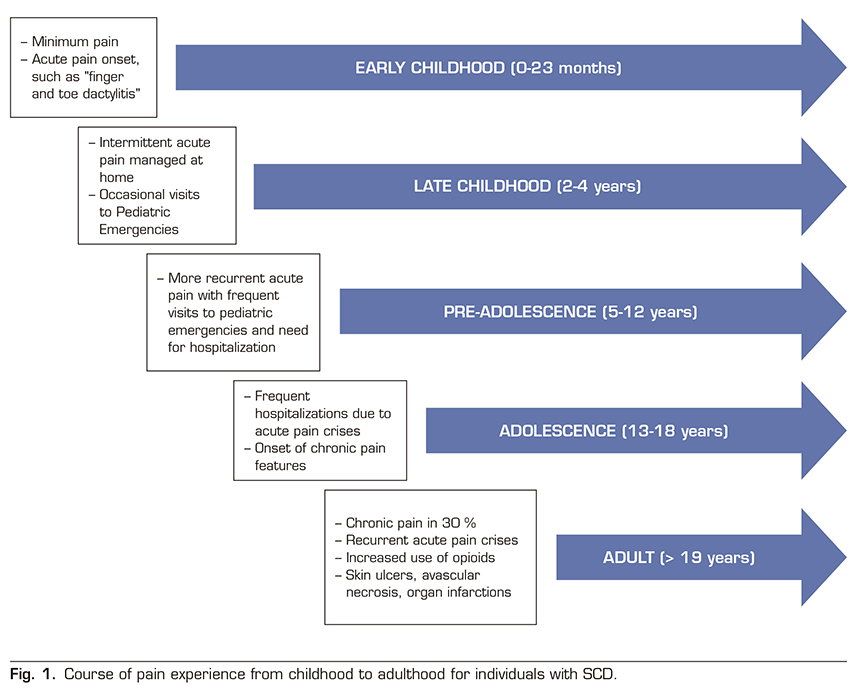
Chronic pain occurs in a significant proportion of patients with SCD, approximately 30%, and increases with age. It has been observed that pain appears from the early ages (0-24 months) in the form of isolated crises and dactylitis phenomena in the hands and feet. Between the age of 2 years and 4 years, most acute episodes are treated at home with intermittent emergency department visits that are increasing between the age of 5-12 years, often requiring hospitalization. In adolescence (13-18 years), acute crisis conditions are varied and there is an increase in hospital admissions, anxiety, depression and sleep disorders appear, and a percentage of patients already meet the chronic pain criteria defined above. In adulthood (> 19 years) these patients present with episodes of acute pain in a context of chronic pain often based on chronic lesions such as skin ulcers, avascular necrosis, or infarcts in various organs and an increased consumption of opioid analgesics (Figure 1) (45). The Pain in Sickle Cell Epidemiology Study (PiSCES) showed that chronic pain occurred in 55% of adults at home in more than half of the days and in 29% of adults in 95% of the days. The etiology of chronic pain in SCD is unclear. However, underlying causes of chronic pain include prolonged hyperalgesia after VOC, organ-specific pain, such as avascular necrosis, and opioid-induced hyperalgesia.
Use of opioids in SCD
The PiSCES project showed that opioids were used during most pain days (78%), and that long-acting opioids and immediate-release opioids were used by 38.8% and 47% of patients, respectively (46). A comprehensive retrospective study on the use of opioids (47) analyzed a total of 3882 patients with a mean age of 17 years (interquartile range [IQR]: 9-36 years). A total of 45 % of the patients were male. Opioid drugs were used by 39.9% of patients, 31.8% using immediate-release drugs only, 0.4% using only long-acting presentations, and 7.8% using both immediate-release and long-acting formulations. The prevalence of opioid use was 8.5% in the 0–9-year age group, increased significantly to 46.3% in the 10–19-year age group (p < 0.0001), and it reached a maximum of 58.3% in the 20–29 age group. Opioid drug doses were converted to oral morphine equivalents (OME). The median daily dose among users was 1.85 mg OME (IQR 0.62-10.68 mg). The opioid dose was 0.54 mg OME per day in the age group 0–9 years, and significantly higher 1.07 mg (p = 0.0002) in the 10–19 years. The age group over 50 years had the highest dose of opioids (6.03 mg OME per day). In non-adult patients (0–17 years) using opioid drugs, the vast majority (87%) used 0-5 mg OME per day, and only 3% used more than 30 mg OME per day. In contrast, 55% of adult patients (over 18 years of age) used 0-5 mg of OME per day, and 23% used more than 30 mg of OME per day. The daily dose of 30 mg OME per day was used as the upper limit in this case because it was a commonly used form of dosage for morphine. Drugs containing hydrocodone and oxycodone were among the most commonly used opioids in both pediatric and adult patients, each accounting for approximately 30% of the total number of opioid prescriptions. Codeine-containing drugs were used more frequently in the pediatric population (27 %) compared with the adult population (4 %). The use of stronger opioid drugs, such as morphine, hydromorphone, methadone and fentanyl, was higher in adult patients (27%) compared with pediatric patients (16%), taking into account body weight adjustment.
Older age, hydroxyurea use, and NSAIDs use were more common among users of high-dose opioids. Hospitalizations and emergency department visits were also more common among high-dose opioid users. High-dose consumers experienced a higher incidence of vaso-occlusive crisis (VOC) (p < 0,0001), acute chest syndrome (ACS) (p = 0.0211), avascular necrosis (AVN) (p < 0.0001), and chronic heart failure (CHF) (p = 0.0027). In multivariate logistic regression models, older patients using hydroxyurea (OR 3.9, 95% CI: 2.8-5.5), and those using NSAIDs (OR 2.0, 95% CI: 1.4-2.7) were significantly more likely to use high doses of opioids. The higher frequency of hospitalizations (more than two admissions per year) was significantly associated with high-dose use (OR 4.7; 95% CI: 3.2 to 6.9) of opioids. The use of NMDA receptor antagonist drugs to avoid the long-term opioid tolerance effect was uncommon. Current recommendations on opioid use in these patients are based on adjusting the expectations of the patients and their families and the goals to be achieved in both acute pain episodes and in the context of chronic pain and understanding SCD course (48). Table 2 summarizes these recommendations.

Depression in SCD
A systematic study showed an estimated prevalence of 26% depression in people with SCD and also a higher prevalence compared to the African American population in general. People with SCD and depression experienced more episodes of pain. In turn, an increase in pain episodes was associated with a higher percentage of depression. Adults with depression had a relative risk of 2.8 times more mental healthcare use than those without depression (49). The PiSCES analyzed 232 SCD patients for depression and anxiety (50). People with depression reported pain in 71% of days compared with 49% reported by people without depression, and people with depression reported increased pain intensity and greater pain interference in their daily activities. Anxiety was also associated with a higher degree of pain. Children with SCD and anxiety disorder had higher pain-related hospital admission rates and longer hospital stays. Adults with anxiety in the PiSCES reported higher mean pain intensity, higher pain-related discomfort, and higher opioid use. These data highlight the reciprocal relationship between mental health conditions and chronic pain in SCD. The long-term effects of treating depression and anxiety on pain will further determine this relationship in the future.
Neuropathic pain in SCD
The diagnosis and treatment of neuropathic pain in individuals with SCD is notably underestimated. Cross-sectional neuropathic pain questionnaires reported by patients have estimated that neuropathic pain occurs in 25-40% of individuals with SCD. Despite data supporting neuropathic pain, systematic strategies for pain management and prevention in these individuals are conspicuously absent. Treatment of neuropathic pain may be difficult due to the difficulty of diagnosis, but may contribute to improving central sensitization of these patients, as many drugs improve the descending pathways of pain. Drugs commonly considered first-line for neuropathic pain treatments include gabapentinoids (gabapentin and pregabalin), serotonin-norepinephrine reuptake inhibitors, and tricyclic antidepressants (51). Limited epidemiological data indicate that medications for neuropathic pain are rarely used in people with SCD. It is not known whether this is due to lack of efficacy data or inadequate diagnosis of neuropathic pain in this population. Infrequent use of neuropathic pain medications was found in two studies that revealed a phenotype of neuropathic pain in people with SCD using patient-reported screening tools. Using the PAINReportIt® tool (a computerized version of Melzack’s McGill Pain Questionnaire [© 1970]), the authors found that 90% of individuals described their pain using words typically associated with neuropathic pain, however, only 5% were using a pain-specific medication (51). In addition, other authors found that 37% of individuals showed evidence of neuropathic pain using the neuropathic examination questionnaire pain DETECT® and only 5% were using a neuropathic pain drug (44). One study analyzed the use of neuropathic pain drugs in the real-world setting in children and adolescents with SCD. Data from the Health Information System of 53,557 inpatient visits were analyzed to determine the prevalence and associated demographics of drugs prescribed for neuropathic pain (gabapentinoids, selective serotonin reuptake inhibitors, tricyclic antidepressants). Data showed that only 2.9% of individuals received a neuropathic pain medication. The chances of receiving a specific drug increased significantly with adults and female sex (52).
Unlike other conditions of neuropathic pain, no systematic reviews of these drugs have been performed in people with SCD due to the limited number of controlled clinical trials. Therefore, data on the use of neuropathic pain medications in people with SCD are scarce. In summary, data from only three controlled clinical trials studying neuropathic pain-focused treatments in individuals with SCD are shown: A phase I open-label study with a neuroleptic drug (trifluoperazine) (53), a randomized controlled pilot trial of pregabalin safety and feasibility in 22 patients (54), and a single-arm phase II study on 3-day transdermal lidocaine (55). Ketamine, a drug that has been shown to be effective in treating some conditions of neuropathic pain and hyperalgesia, is being outlined as a possible treatment for the pain of the disease. Limited data on the use of ketamine for the treatment of acute pain (VOC) in SCD suggest that this drug may be effective (56, 57), but additional studies are needed.
CONCLUSIONS
Pain is the most common complication of SCD and the leading cause of hospitalization in affected people. Acute pain episodes are also an independent predictor of mortality and there is an evident transition from acute episodes in early age to chronic pain in at least 30% of adult patients. The pathophysiology of pain is complex and has been attributed to several biological factors, including oxidative stress, vaso-occlusion, ischemia-reperfusion injury, local inflammation and finally a process of neuro-inflammation and central sensitization leading to chronic pain syndrome. Despite this complex biology, painful events requiring hospitalization are simplistically called “episodes of acute vaso-occlusive pain” by the community of clinicians, and we should not be tempted to stay in this superficial view. Because in the background lies a complex group of clinical signs and symptoms leading to severe physical and mental involvement in these patients requiring a multidisciplinary biopsychosocial approach (Figure 2).
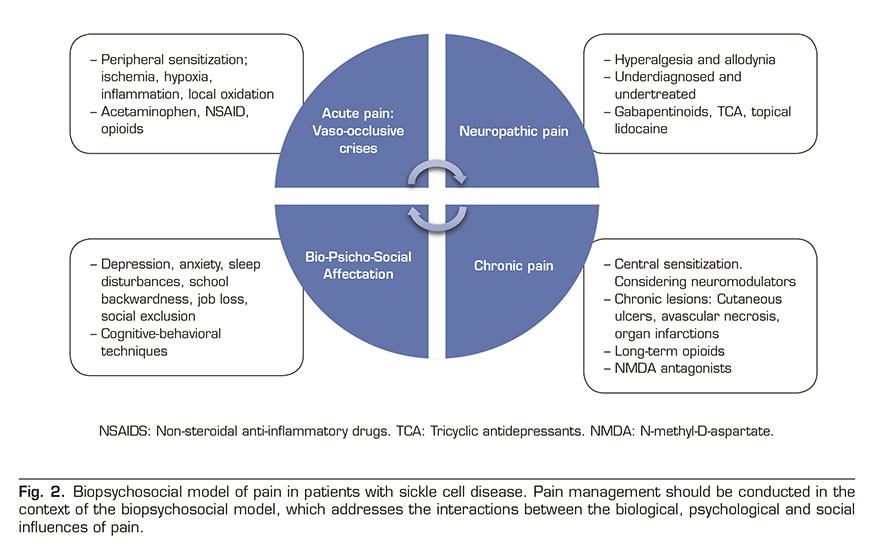
Neuropathic pain is an emerging phenotype within the painful spectrum of SCD. It is caused by a somatosensory nervous system injury or disease and has been estimated to occur in approximately 25-40% of adolescents and adults with SCD. Diagnostic modalities for neuropathic pain, including validated questionnaires including pain descriptors, quantitative sensory tests, and functional neuroimaging, have been evaluated in small cross-sectional studies in adolescents and adults with SCD. However, these diagnostic tests are not currently used in routine patient care. Age, female sex, and hydroxyurea use have been reported to be positively associated with neuropathic pain in SCD, although definitive risk factors for pain prevention in this population have not been identified. A few early-stage studies have begun to investigate both neuropathic pain and central sensitization with neuromodulator agents (ketamine, pregabalin, lidocaine) in individuals with SCD. However, evidence-based strategies for treating pain in sickle cell disease are scarce, and existing literature suggests that specific treatments for neuropathic pain are rarely used, and anti-inflammatory drugs and opioids remain first-line drugs, and therefore, they are overused.
CONFLICT OF INTEREST
None of the authors has a conflict of interest.
REFERENCES